
Microbial Community Profiling:
We offer services to profile a range of microbial community compositions:

- Standard 16SV4 - Our standard 16SV4 protocol (Kozich et al., 2013) targets and amplifies the V4 region of bacterial and archaeal 16S rRNA-encoding genes.
- 16S EMP - A modified set of primers by the Earth Microbiome Project (EMP) that also targets the V4 region but with added degeneracies to be more inclusive of bacterial and archaeal species found in some environments (Walters et al., 2015).
- Fungal ITS - Primers targeting the fungal Internally Transcribed Spacer (ITS) region for amplification of a broad spectrum of fungal taxa in complex microbial communities. (Taylor et al., 2016). Library generation methods are available for characterizing the ITS1 and ITS2 regions.
Community profiling can occur on purified DNA samples from a variety of sources, including blood, stool, tissue, saliva and soil. We accept samples in a 96-well plate format, with up to 94 samples and 2 blank wells per controls. Purified nucleic acids can be quantified at the Core, or we will process the samples as described in our Nucleic Acid Extraction service. Libraries are cleaned, normalized, and undergo quality control before sequencing on the Illumina MiSeq platform. Clients have the option to run combined community analysis for characterizing fungal and bacterial composition in their sample sets. Output data (ex: FASTQ files) can be returned to our clients via Illumina’s BaseSpace Sequence Hub or on a USB flash drive.
If you are ready to submit samples for community profiling, we process all internal submissions through our MiCores website and external submissions through our Qualtrics survey.
To learn more, please refer to the Frequently Asked Questions below, or reach out to us at microbiomecore@umich.edu.
Submission/Pricing
1. If some of my samples fail to amplify can the lab try other conditions to improve amplification?
Yes, we will ask whether you would like us to troubleshoot your samples. Additional charges apply. Also, you have the option to preselect one round of troubleshooting for up to 20 samples.
2. Are there protocols I can follow to prepare my samples for submission/shipping?
Yes, please refer to the relevant service’s page for links to detailed instructions.
3. What is the process for an internal or external submission?
Internal clients can login to your MiCores account and fill out a service request. External clients are asked to use our Qualtrics survey.
4. How do I get a MiCores account?
Please consult this User Help Guide to sign up for a MiCores account.
5. How long will you keep my DNA (submitted or extracted)?
We will keep DNA for 60 days after submission; samples will be disposed after this period. If you are an external client and have sent in samples for DNA extraction, we will ship your DNA back at cost to you. For internal clients, please pick up DNA when receiving your USB containing the data. We are not responsible for DNA that was not claimed and disposed after the grace period.
It is best practice to submit an aliquot of your DNA for submission and keep a set for yourself.
6. Can I use your services if I am not affiliated with the University of Michigan?
Yes, we do take external clients. There is a surcharge for external clients.
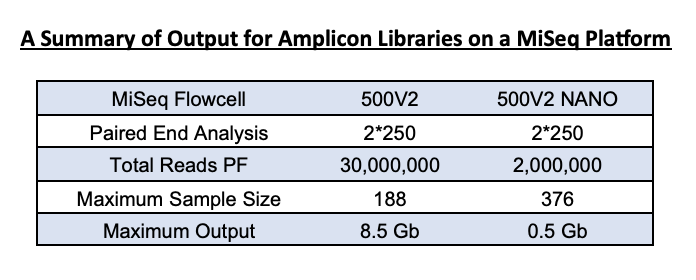
7. What is the Illumina Nano Kit?
The MiSeq Nano kit is a smaller version of the full 500-cycle Illumina kit we use for 16S rRNA gene sequencing. It yields fewer reads (~1.5 – 2 Million reads vs the full 500v2 kit’s estimated 20 Million reads). This output is enough for community analysis for a 1-2 plate sample set.
8. Do you have a template for the plate map I need to submit electronically?
Yes, please find plate map in downloads below.
9. Why am I asked if I do metabolic mouse work on the submission form?
We are part of the National Mouse Metabolic Phenotyping Centers (MMPC). The MMPC is a NIH-sponsored resource that provides experimental testing services to scientists studying diabetes, obesity, diabetic complications, and other metabolic diseases in mice. By initiating your order through the MMPC, your submission ensures continued financial support for the Core services our lab provides.
Nucleic Acid Extraction
1. Do I have to add my samples to the Bead Plate or do you do it?
We do not add samples to the Bead Plate. We feel it is best that the client adds the samples as this reduces the risk of error.
2. How do I get a Bead Plate?
For our internal clients, Instructions can be found in our MiCores service request forms (specifically in ‘Community Analysis Submission’ or ‘Stand-alone DNA Isolation Service’).
For our external clients, we will ship bead plates upon confirmation of project submission.
3. How much sample should I add to each well of the Bead Plate?
250 uL or 0.25 g can be added to each well. Please seal properly with the silicone mat provided before returning to the Core. If we are to run community analysis on the extracted sample set, we ask for clients to have three empty wells as controls for downstream processing.
4. Where should I add my samples to the Bead Plate?
A biohazard hood using appropriate personal protective equipment.
5. I will be shipping my bead plate to the core, are their guideline available for me to follow?
Yes! Please use our shipping guide to learn more about how to appropriately prepare bead plates for shipping. We recommend shipping the samples already frozen in the bead plates as the package will undergo some handling during the transit process. Also, do reach out to the core (microbiomecore@umich.edu) to share delivery details so we may track the package on our end. We do notify clients on delivery of their packages, so please ensure to include contact information within the package.
Community Analysis
1. What are your average reads per sample?
For samples that have amplified well, the average is 10-20K per sample on the nano 500v2 flow cell. A standard 500V2 flow cell has an output of 30 million reads, and so for a 384-sample set, you can expect an output of 40-60K per sample. However, this cannot be guaranteed as many factors can influence average reads/sample.
2. Do I have to pay for an entire plate if I am submitting less?
Yes, if you do not have an entire plate, you are still responsible for the cost of the entire plate. On the plus side, you will receive more coverage of those samples than if you submitted a full plate. We cannot coordinate submissions, however, we offer internal clients a project matching board, where they can reach out to other potential clients who may have a partial set of samples for submission.
3. Should I quantify my DNA?
It is not necessary but recommended. If quantifying the sample, please use a fluorescence-based assay such as Picogreen since it will provide more accurate quantification measurements. Nanodrop is not recommended. Quantification results are helpful for us to determine template volumes required when setting up a library. We do offer DNA quantification as a add on service when setting up ITS or 16SV4 libraries, however if we are carrying out DNA extraction, it is already built into our services.
4. What is touchdown PCR and how does it differ from standard PCR?
Touchdown PCR is offered for generating 16S V4 libraries, and usually applied for low biomass samples. The PCR program is modified from our standard PCR program, allowing for less stringent amplification over the first 20 cycles with an initial annealing temperature higher than the optimal Tm of the primers. This temperature is gradually reduced over the first 20 cycles until the Tm temperature is reached. This increases sensitivity in PCR amplification as described here. Additionally, after 20 rounds of touchdown PCR, another 20 rounds of PCR are performed at optimal annealing temperature.
Touchdown is a great tool for low biomass samples but should be used with caution as any trace amounts of 16S will amplify including that in the negative controls.
5. For fungal community analysis, which ITS profiling method should I use?
We offer both ITS1 and ITS2 community profiling services. It is recommended that our clients determine which library type is better suited to their community group. However, the ITS1 region is comprehensive and provides generally higher quality sequencing results. The ITS2 library type provides more specificity for certain community groups. The sequencing quality for ITS2 may differ by community type, and in some instances read 2 results are of low quality. Some clients prefer ITS2 profiling since it provides more definition in their analysis and may choose to use the forward read alone for analysis.
6. Can you combine library types on a sequencing run?
Yes, a combination of 16SV4 and ITS libraries can be run together. We can combine a total of 4 plates prepared for either library on a standard 500V2 run (4 * 96 samples run of unique dual indexed samples) or a two plate combination (2 * 96 samples run of unique dual indexed samples) on a nano 500V2 run.
7. What are the stages involved in processing my project for community analysis?
We receive purified DNA from clients or can extract DNA samples at the core. Samples undergo a primary round of PCR where library amplicons are uniquely barcoded through dual indexing. For successful amplification, variables such as sample concentration, library purity, library generation type are taken into consideration. We aim to get all samples to amplify through an automated PCR setup. The client has the option to troubleshoot any samples that remain unamplified. Once the library is completed, samples are normalized and pooled per plate. Library pools undergo QC before they are setup to run for sequencing. Our turnover times may vary based on the volume of projects being processed at the core. We aim to provide quick turnover of submissions yet at times processing can take up to 3-4 weeks. If you would like to follow up with us about an ongoing project, please do reach out us, microbiomecore@umich.edu.
Data
1. What is the Mock control and why do I need it?
The mock community used is commercially produced ZymoBIOMICS Microbial Community DNA Standard (cat# D6306), which is a mixture of genomic DNA extracted from pure cultures of eight bacterial and two fungal strains. This mock community can be used for error analysis (see: Mothur Wiki). The FASTA file for this mock can be found in a downloadable file below.
2. What type of data will I receive?
We return FASTQ files. Additionally, we provide certain run metrics, which include % reads / samples pass filter.
3. Are the indices removed from the FASTQ files?
We do not prompt adapter trimming on the FASTQ results. The 16SV4 region in most cases within the 250 base pair reads taken, and so none of the remaining adapter region such as barcodes gets included in the results. ITS results are more variable lengths, and so reads may require adapter removal.
4. How do I receive the data?
All sequencing project data can be transferred on Illumina’s BaseSpace website. For an additional cost, you may request the data also be transferred to a USB.
5. How do I open a Basespace account?
Illumina’s BaseSpace Sequence Hub account is available without subscription and can be used to store up to 1TB of data. The account expires if not used in the last 6 months. You do not need any institutional affiliation to setup an account. Additional tools available through paid subscription are not necessary if using alternative pipelines are used for data analysis, such as MOTHUR for 16SV4 or for ITS community analysis. You may sign up for an account here: https://www.illumina.com/products/by-type/informatics-products/basespace...
6. How do I access my data?
Illumina’s BaseSpace Sequence Hub accounts are linked to your email and this email address is used by the core to transfer your data results. Signing into your account, on the top right corner of the dashboard is the notifications bell icon. Any pending data transfers from the core will be found here. Two ownerships of transfers are sent by the core, which include the run metric results and project results. Once you have accepted ownership of these, the core loses access to the data.
7. How do I download FASTQ results off Basespace?
There are two methods available for downloading FASTQ files. One is directly from the website or in instances that website slows down due to traffic, it is quicker through Illumina’s Command Line Tool.
a) Website Download – FASTQ files are found under the samples tab and you can download the whole set or a portion of files at a time > you can download these by navigating to either the runs or project tab on the top most menu of BaseSpace’s dashboard> click on the run you would like to access > choose the samples tab > select the files of interest > hit downloads under file icon. You will be prompted through the steps to carry out the download of selected files.
b) Illumina has their BaseSpace CLI tool for quick download of mass files, especially FASTQ. Super helpful when the site slows down due to traffic. Here is a general overview of BaseSpace CLI: https://developer.basespace.illumina.com/docs/content/documentation/cli/.... You'll be able to download BaseSpace CLI using links from that document. Also, here's a video that goes over the download process: https://www.youtube.com/watch?v=RQKhGcQf3Vo.
8. How can I transition to the free Basespace account?
If you are looking to transition to the free account, you can transfer your files to your personal settings instead of the enterprise account. The basic account is readily available to people outside of institutional settings. You will find that you have both settings currently available to you. You can change the settings to accept uploads to the personal account.
• Top right of page under your account pulldown menu > choose settings > Under default location for uploaded runs > choose personal
2) Transferring sequencing runs to the same email address. Two transfers are needed to transfer the run info and project results for each sequencing run
• Under the Runs tab on top menu > click on project > choose the share icon > transfer ownership > use the same account email
• Under the Project tab on top menu > click on bulletin for project > choose the share icon > transfer ownership > use the same account email
You will have access to your runs under notifications, and runs will be available to you when you sign in the next time you log in. You may find this Illumina bulletin helpful: https://knowledge.illumina.com/software/cloud-software/software-cloud-so....
6. What if I lose my data?
We keep backups of data for six months so if you lose your USB or delete your BaseSpace run within this time frame, we will likely have a backup copy. A fee of $75 will be charged to retrieve your data.
7. What is in the workbook?
The standard workbook contains a methods sheet, your plate map, the PCR conditions for each of your samples, any troubleshooting conditions, post-PCR QC results, a summary of run metrics, and how well your samples indexed.
8. How do most of your clients analyze their 16S rDNA data?
Most of our clients use Mothur to analyze their data. The MiSeq SOP on the Mothur wiki page is an excellent resource. Additionally, Pat Schloss offers numerous workshops throughout the year.
9. Do you provide a service to analyze the data?
We are looking to onboard analytical support for 16SV4 community analysis projects processed at the core. Please reach out to our core (microbiomecore@umich.edu), if interested.
10. Citing the Microbiome Core
We ask that all publications and presentations of research results supported by the Microbiome Core contain the following (or equivalent) acknowledgement: "This research was supported by work performed by The University of Michigan Microbiome Core"
Please let us know as soon as you have an article accepted for publication, poster, or meeting presentation which has received Microbiome Core support. Early notification helps us to track the activities being supported by the Microbiome Core. Submit your notifications by email to microbiomecore@umich.edu. Be sure to include a copy of the presentation abstract or complete accepted article.