

We offer two services for sequencing of bacterial genomes on the Illumina MiSeq Platform:
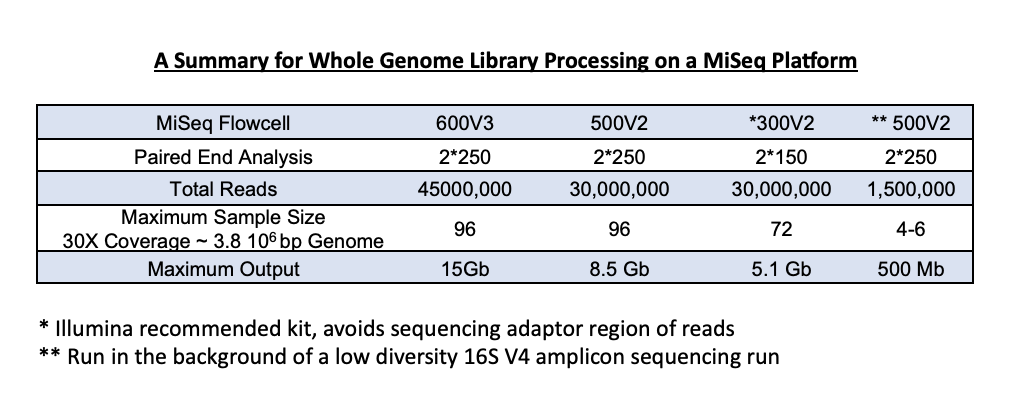
1) High-throughput - Ideal for larger sample sets of up to 96, sequencing is carried out on an individual reagent cartridge. We have multiple sequencing kits available depending on the number of samples and desired coverage. Optimal downstream analysis, based on a 3.87Mb bacterial genome, calls for about 30X coverage. We can adjust the run to be sure you get the coverage required, based on the size of your genome.
2) Low-throughput – This option can be useful for preliminary analysis for those interested in pursuing a larger project. You may choose submission of 6 or 24 samples. Again, samples are loaded to ensure at least 30X coverage, based on the size of the genomes.
Processing is done using the Illumina DNA Prep Kit, (Cat: 20018704 / 20018705) to create unique dual-indexed sequences.
Purified DNA can be sent to the core or we can perform extraction of DNA from microbial cultures using the Qiagen Microbial DNA kit (27200-4). If you are submitting extracted DNA, we ask that you provide concentrations using a fluorescence-based detection system, such as picogreen or Qubit. We would also ask for a concentration of at least 5 ng/µL, in a volume no less than 20 µL. If we are performing the extraction, then quantification is included.
If you are ready to submit samples for microbial genome sequencing, we process all internal submissions through our MiCores website and external submissions through our Qualtrics survey.
iLabs Submission/Pricing
1. Is there a form to fill out prior to submitting samples for sequencing?
Yes, please login to your MiCores account and fill out a service request.
2. How do I get a MiCores account?
Please consult this User Help Guide to sign up for a MiCores account.
3. Can I use your services if I am not affiliated with the University of Michigan?
Yes, we do take external clients. There is a surcharge for external clients.
4. Where can I find pricing details?
Upon entering our MiCores account, you can find pricing for services in the "Request Services" tab, in the second half of the page, listed under "Service Price List".
Microbial Whole Genome Sequencing
1. Can the core extract my DNA?
Yes, we often utilize Qiagen’s MagAttract Microbial DNA kit for samples stemming from culture media, in sets of 96. We can also just do the extraction and DNA normalization part of the whole genome sequencing project.
2. What size of genomes do we process?
The kit can accommodate smaller plasmids to large microbial genomes of any size.
3. What concentrations are required?
For processing genomes, we ask that the client provide around 20uL of sample at 5ng/uL. For setting up library generation reaction, we use an input amount between 1-24ng in a 30uL volume. Extra sample may be needed for quantification and if troubleshooting is required.
4. Which kit is used for library preparation?
We use the Illumina DNA Prep Kit, which utilizes bead-linked transposomes for tagmentation, generating dual indexed samples. The kit generates fragments of various lengths, with an average insert size of 350 basepairs.
5. What is the difference between our low-throughput and high-throughput service?
Our high-throughput service is appropriate for large sample sets of up to 96. An additional cost of the sequencing kit is added based on the clients need for coverage and the number of samples processed.
Library generation occurs in the same way for the low-throughput genome service; however, these samples are run in the background of a 16S sequencing run. We accept up to 24 samples and can get around 30X coverage from these background genome runs.
6. How is coverage determined?
Coverage (C) is dependant on the size of the genome being sequenced (G), number of reads generated (N) and the read length (L) of the samples run.
C=L*N /G
We load our background genomes at 13% of the total load amount, generating about 1-2 million clusters per sample set. A sample set of 5 genomes in the background, around 3.8 * 106 basepairs, will provide 30X coverage for each.
7. If some of my samples fail to amplify, then can the lab try other conditions to improve amplification?
Yes, we will ask whether you would like us to troubleshoot your samples. Additional charges apply.
8. How long will you keep my DNA (submitted or extracted)?
We will keep DNA for 60 days after submission; samples will be disposed after this period. If you are an external client and have sent in samples for DNA extraction, we will ship your DNA back at cost to you. For internal clients, please pick up DNA when receiving your USB containing the data. We are not responsible for DNA that was not claimed and disposed after the grace period.
It is best practice to submit an aliquot of your DNA for submission and keep a set for yourself.
Data
1. What will I receive after my project has been completed?
Upon completion of the project, the client receives project details such as indexing barcodes, the number of reads per sample, and estimated coverage along with their FASTQ files. Most clients have preferred pipelines for data analysis, however if you are looking to get some assistance, the BCRF Bioinformatics Core is a good place to start. iki). The FASTA file for this mock can be found in a downloadable file below.
3. Are the indices removed from the FASTQ files?
Yes.
6. What if I lose my data?
We keep backups of data for six months so if you lose your USB or delete your BaseSpace run within this time frame, we will likely have a backup copy. A fee of $75 will be charged to retrieve your data.
7. What is in the workbook?
The standard workbook contains a methods sheet, your plate map, the PCR conditions for each of your samples, any troubleshooting conditions, post-PCR QC results, a summary of run metrics, and how well your samples indexed.
Citing the Microbiome Core
We ask that all publications and presentations of research results supported by the Microbiome Core contain the following (or equivalent) acknowledgement: "This research was supported by work performed by The University of Michigan Microbiome Core"
Please let us know as soon as you have an article accepted for publication, poster, or meeting presentation which has received Microbiome Core support. Early notification helps us to track the activities being supported by the Microbiome Core. Submit your notifications by email to microbiomecore@umich.edu. Be sure to include a copy of the presentation abstract or complete accepted article.